Mehr als 15 Gene sind für das Noonan-Syndrom bekannt. Die größten Anteile bei den Fällen mit nachgewiesener Mutation (Genveränderung) entfallen auf die Gene PTPN11 (40-50%), SOS1 (10-15%), RIT1 (5-10%) und RAF1 (5-8%). 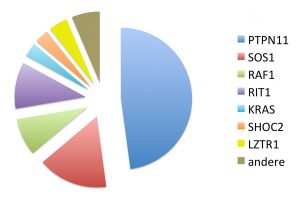
Unterschiede in der Ausprägung der Erkrankung hängen zum Teil mit der spezifischen Genveränderung zusammen. Zusammenhänge zwischen Genmutation und klinischer Ausprägung (Genotyp-Phänotyp-Korrelation) sind in der Tabelle dargestellt:
Weitere Informationen
Genname | Anteil bei Noonan-Syndrom | Klinische Merkmale / Besonderheiten |
PTPN11 | 40-50% | „klassisches“ Noonan-Syndrom, häufig Pulmonalstenose, relativ selten hypertrophe Kardiomyopathie spezifische Mutationen (p.C279Y, p.T468M u.a.) sind für den Untertyp Noonan-Syndrom mit multiplen Lentigines (LEOPARD-Syndrom) verantwortlich |
SOS1 | 10-15% | geringere Häufigkeit von Kleinwuchs und kognitiven Defiziten, ausgeprägtere Anomalien der Haut und Haare, Ptosis, Neigung zu Riesenzelltumoren des Kiefers |
RIT1 | 5-10% | geringere Häufigkeit von Kleinwuchs und kognitiven Defiziten, Herzanomalien in >90%, darunter auch hypertrophe Kardiomyopathie (60%), häufiger Lymphödeme |
RAF1 | 5-8% | hypertrophe Kardiomyopathie (>80%), auch schwere Formen der hypertrophen Kardiomyopathie, Makrozephalie, multiple Pigmentnävi |
KRAS | 2-3% | variable Ausprägung mit Übergang zwischen Noonan- und CFC-Syndrom, häufiger kognitive Einschränkungen |
LZTR1 | Noonan-Syndrom (1-5%) | variable und manchmal sehr milde Ausprägung in Fällen mit dominanter Vererbung, rezessive Vererbung möglich |
NRAS | ca. 1% | zu geringe Patientenzahl für statistische Analyse, |
RRAS | <1% | zu geringe Patientenzahl für statistische Analyse |
RRAS2 | <1% | zu geringe Patientenzahl für statistische Analyse |
MRAS | <1% | nur wenige Fälle beschrieben, häufig hypertrophe Kardiomyopathie |
SOS2 | ca. 1% | relativ geringe Patientenzahl, wahrscheinlich ähnlich SOS1, häufig Lymphödeme |
CBL | ca. 1% | Noonan-ähnlicher bis unspezifischer Phänotyp, Prädisposition für JMML, Gefäßveränderungen |
SHOC2 | 2-3% | typische Veränderungen der Haare („loses Anagenhaar“), dunkle Pigmentierung, Herzfehlerspektrum, Wachstumshormonmangel |
PPP1CB | <1% | ähnlich wie Betroffene mit SHOC2-bedingtem Noonan-Syndrom |
BRAF | ca. 1% | einzelne Fälle, häufiger Entwicklungsdefizite, häufiger Anomalien der Haut und Haaren, die meisten Träger von BRAF-Mutationen haben die Diagnose CFC-Syndrom |
MAP2K1 (MEK1) | <1% | einzelne Fälle, häufiger Entwicklungsdefizite, die meisten Träger von MAP2K1-Mutationen haben die Diagnose CFC-Syndrom |
MAP2K2 (MEK2) | <1% | einzelne Fälle, häufiger Entwicklungsdefizite, die meisten Träger von MAP2K2-Mutationen haben die Diagnose CFC-Syndrom |
Das Noonan-Syndrom ist aus biologischer Sicht eine Störung des RAS-Signalwegs (auch RAS-MAPK-Signalweg genannt). Dieser ist in praktisch jeder Körperzelle vorhanden und dient der Übertragung von Signalen, wie beispielsweise der Signale von außen auf die Zelle auftreffender Botenstoffe (Hormone, Wachstumsfaktoren), zum Zellkern. Viele Vorgänge des Wachstums, der Differenzierung und Reifung von Zellen und Geweben werden so gesteuert. Die Biologie bedient sich für diese Signalübertragung einer Kette von Aktivierungsschritten miteinander verbundener Signalproteine. 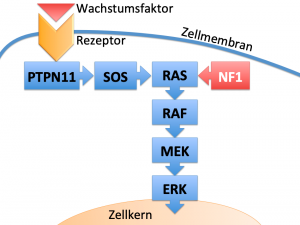 Das Gleichgewicht zwischen RAS-Aktivierung und -Inaktivierung ist durch verschiedene weitere Moleküle fein reguliert, diese genaue Steuerung ist wichtig für die ordnungsgemäße Funktion des Systems.
Das Gleichgewicht zwischen RAS-Aktivierung und -Inaktivierung ist durch verschiedene weitere Moleküle fein reguliert, diese genaue Steuerung ist wichtig für die ordnungsgemäße Funktion des Systems.
Bei den Mutationen (Genveränderungen), die zum Noonan-Syndrom und verwandten Erkrankungen führen, handelt es sich im eigentlichen Sinne nicht um Defekte, die den Ausfall der Genfunktion bewirken. Vielmehr führen sie zu einer Fehlsteuerung und Überaktivierung des RAS-MAPK-Signalwegs. Über welche Mechanismen diese Fehlsteuerung in den Zellen aber zu den einzelnen, für das Noonan-Syndrom typischen Veränderungen in Organen und im Körper führt, ist bis heute noch nicht hinreichend genau erforscht.
Das Noonan-Syndrom ist eine genetische und prinzipiell vererbbare Erkrankung. Der Vererbungsweg ist im Allgemeinen autosomal-dominant. Das bedeutet, dass Kinder von Betroffenen mit einer Wahrscheinlichkeit von 50% die ursächliche Genveränderung und damit die Erkrankung erben. Der größere Teil (etwa 80%) der Kinder mit nachgewiesenem Noonan-Syndrom hat aber keinen betroffenen Elternteil, weil es sich um eine zufällig neu entstandene Genveränderung (Neumutation) handelt.
In den Fällen, wo das betroffene Kind eine Neumutation hat, ist das Risiko für die Wiederholung eines Noonan-Syndroms bei einem weiteren Kind dieser Eltern nur geringfügig erhöht.
Es gibt ausnahmsweise auch Fälle mit autosomal-rezessiver Vererbung des Noonan-Syndroms. Bei dieser Art der Vererbung können gesunde Eltern, die beide Träger einer Genvariante sind, mehrere betroffene Kinder haben.
Für Fragen zu individuellen Wiederholungsrisiken und zu Möglichkeiten einer vorgeburtlichen Diagnose ist eine genetische Beratung zu empfehlen.